
The butterfly effect
Skin too fragile to touch
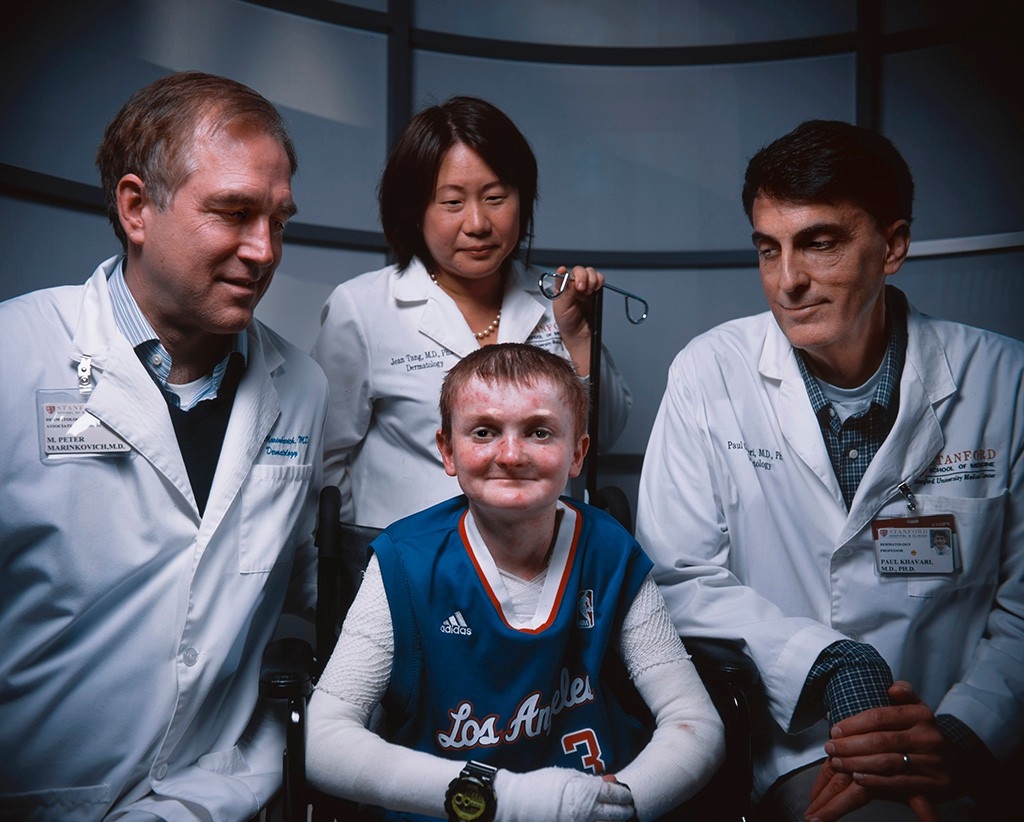
Jan. 25, 1997, was part of dermatologist Paul Khavari ’s first weekend on call at Stanford. He remembers it clearly. Very early that Saturday morning, when it was still dark, he admitted a newborn from Gilroy with a horrifying constellation of symptoms.
Extra:
Related video: The butterfly effect
“His whole body, his skin was blistered and falling off everywhere someone had touched him,” Khavari remembers. “His parents were devastated, of course, at a time that was supposed to be one of the most joyful of their lives.”
The baby had a severe form of a blistering skin disease called epidermolysis bullosa, and doctors like Khavari, now the chief of dermatology at Stanford, knew that his life span would be measured in years of nearly unimaginable pain. As he and his colleagues gently outlined the disease to his parents, the baby’s father began to faint. Doctors rushed to support him.
“That was such a heart-wrenching evening,” the baby’s mother, Lorraine Montello, recalls. She and her husband had four other healthy kids and had expected an uneventful birth and recovery. Instead, their baby, Garrett Spaulding, was immediately transferred to Lucile Packard Children’s Hospital Stanford. They followed as soon as Montello was discharged.
“It was a terribly stormy night,” recalls Montello. “Garrett had to be transported by ambulance because the weather was too rough to fly. We rushed into the room in the middle of the night to see a group of three very tall doctors standing over him. I thought, ‘This just doesn’t look good.’”
The doctors — the then-chair of the dermatology department, Al Lane, MD; the director of Stanford’s Blistering Disease Clinic, Peter Marinkovich, MD; and Khavari — were clustered around Garrett’s bed. They were some of the best-trained dermatologists in the country, but even they could offer no more than pain relief and bandaging to the wailing newborn.
“They were three giants, leaning over my tiny baby,” recalls Montello wryly, noting that each of the doctors stands well over 6 1/2 feet tall. “That was such an awful, horrible and depressing evening, but that provided a tiny bit of comic relief.”
Montello had no way of knowing at the time, but the trio of doctors were also well on their way to becoming giants in the study of epidermolysis bullosa, which affects only about 20 infants per 1 million births in the United States. (About two of those 20 have the same form of the condition as Garrett has.) Lane and Marinkovich had already devoted years of their lives to learning the molecular causes of EB, as it’s come to be called. And Khavari had just published a paper describing ways to apply then-nascent gene therapy techniques to correct genetic skin defects like EB.
The doctors’ single-minded devotion was the product of the previous decade of study at Stanford focusing on the rare disease, initiated by dermatologist Eugene Bauer, MD, in 1988. Now, nearly 30 years later, the Stanford team has launched the world’s first stem-cell-based clinical trial aimed at replacing the defective gene in patients’ skin cells with a working copy. The researchers have used sheets of corrected skin cells from four patients to cover some of their wounds; preliminary results will be announced this summer.

If the treatment is deemed safe, the physicians will move to a larger trial in the hopes that one day no child will have to suffer like Garrett.
“Of all the skin conditions we see in dermatology, EB is the most completely debilitating and awful,” says Jean Tang, MD, PhD, an associate professor of dermatology and who, along with Marinkovich, directs the phase-1 clinical trial. “If we could cure only one condition in this field, it would be EB, hands down. I don’t know any dermatologist who would answer differently. It’s hard for healthy people to realize just how important your skin is and how limited EB patients’ lives are.”
Patients with severe forms of EB experience the equivalent of third-degree burns, as their skin blisters and sloughs off at the slightest friction. A parent’s touch. A friendly handshake. A step in a snug sneaker. In some circles they’re known as the “butterfly children” to compare the fragility of their skin to that of a butterfly’s wing. In the Netherlands, at least two infants with the most severe forms of the disorder have been euthanized to end what’s described as unbearable, unrelenting pain.
There is no cure.
The condition demands extensive bandaging, from head to toe, in a regimen that can take hours every morning and evening. Infection of the open wounds is common, and fingers and toes frequently fuse together as a result of the constant scarring and healing between the digits. Because the condition can also affect internal organs and the gastrointestinal tract, patients struggle to maintain adequate nutrition and often experience organ and dental problems.
Garrett Spaulding is now 18. Montello, who underwent a crash course in the disease after his birth, has served for the past decade as coordinator for Stanford’s EB clinic. She spends about five hours every other day replacing the gauze that covers much of Spaulding’s body. “A large percentage of his body is covered with deep, open wounds,” she says. She often has to take breaks from the bandaging because of the excruciating pain it causes him. “When he was younger he’d say, ‘Mom, I hate you, I want to die’ — and he meant it,” she recalls.
“The amount of suffering is astronomical,” says Khavari. “It’s intimidating. I remember thinking, that night in 1997, ‘We need to be able to do more for these children.’”
In the intervening years, Khavari and his colleagues have dedicated themselves to understanding the disease and finding new ways to treat it.
“It’s been a decades-long journey,” says Khavari, but they haven’t wavered. The ongoing clinical trial focuses on replacing the defective gene in stem cells in the skin that develop into keratinocytes — the cells that overlap like shingles to form a protective barrier against the outside world. Healthy keratinocytes produce a protein called type-7 collagen that is essential to hold skin layers together. People with Spaulding’s form of EB have a mutation in the gene that encodes this protein. The researchers correct the gene in naturally occurring stem cells from the patients and allow them to grow into thin sheets of skin to patch onto the patient’s wounds, a process known as grafting.
In addition, the group is pursuing at least two other approaches to combat the disease. One, called therapeutic reprogramming, is similar to the current trial but would rely on stem cells created in a laboratory. Another, called protein therapy, would directly deliver the missing collagen protein to the surface of the skin.
By covering all the bases, the researchers hope to find at least one approach, or perhaps a combination of techniques, that can help people like Spaulding.
“We are at a unique moment in history,” says Anthony Oro, MD, PhD, a professor of dermatology at Stanford and a member of the EB team. “We have access to stem cell biology, gene-editing tools and bioengineering and transplant biology. We’re hoping that this will all crystalize in better ways to treat these kids.”
Khavari sums up the dedication of the group more simply: “We want to make these children well.”
The skin is the largest organ in the body. In an adult human, the skin totals about 1.5 to 2 square meters, or roughly the area of a nice-sized throw blanket. It serves as an essential barrier between the messy gloop and rippling red cords of our innards and muscles and a world teeming with dangers — infectious microbes, toxic substances and even the hot and cold air around us that would quickly desiccate us into shriveled husks if we were left unprotected. It allows us to feel changes in temperature and pressure, and, yes, even pain. One square centimeter of human skin contains 200 pain receptors, all primed to transmit pain signals in the most efficient, speedy way possible to encourage us to lift our hand off that burner, or to recoil from the knife that has only begun to break the skin. In EB patients, those receptors are firing almost nonstop.
“We don’t know what it is like to not be in pain,” says Paul Martinez, a 32-year-old man with EB from Stockton, California. Martinez is a participant in Stanford’s current clinical trial. “It’s just normal for us.”
The top layer of the skin, the epidermis, is made up primarily of keratinocytes. These flat, irregularly shaped cells create an impervious sheet locking out water and bacteria. Under the epidermis, which can range from 0.2 to 4 millimeters in thickness, are two fibrous sheets collectively called the basement membrane. Like the bologna in a sandwich, the basement membrane separates the epidermis from the dermis, which consists of connective tissue housing hair follicles, sweat glands and blood vessels.
The basement membrane is the culprit in EB. People with Spaulding’s form of EB have mutations in the gene for type-7 collagen — one of the most important proteins you’ve never heard of. Like rebar used to stabilize layers of concrete in building, or toothpicks holding together that slippery sandwich, type-7 collagen connects and anchors the dermis to the epidermis to allow them to move smoothly together in response to pressure or friction. In its absence, the layers slide past one another, causing damage and blistering at the slightest friction or pressure. Often the blisters grow and migrate across the skin and eventually pop, sloughing off skin and leaving an open, weeping wound that heals slowly if at all.
This cycle of blistering and healing often eventually causes the toes and fingers of patients to fuse together and curl inward, leading to a condition called “mitten hand” that resembles a ball of flesh with no digits. Surgery can sometimes free the hand, but the condition inevitably recurs.

In decades past, children with EB died within a few years from massive infection or fluid loss from their wounds. They are often severely underweight, as their bodies frantically divert calories to skin healing, rather than using them for growth or weight gain. As bandaging techniques and antibiotic therapies have improved, children are living into their 20s, only to face another challenge: The constant rounds of regeneration drastically increase the chances of developing a skin cancer known as squamous cell carcinoma. Although this type of cancer is often curable in healthy people, it recurs repeatedly at multiple sites across the body in people with EB. The cancer, combined with frequent difficulties with nutrition as patients struggle to eat with painful sores in their mouths and throughout their digestive system, as well as other organ problems, means that few with EB live past the age of 30.
In this context, Martinez is an old man.
Despite the severity of EB, there are some hopeful aspects of the disease. Although type-7 collagen is encoded by a large, unwieldy gene, it’s the only culprit in the form of EB that Spaulding and Martinez have. The fact that it affects the skin also means the damage is accessible to physicians and any changes can, for the most part, be monitored visually.
“The skin is a very accessible organ,” says Zurab Siprashvili, PhD, a senior scientist in Khavari’s laboratory. “Compared with the liver or heart, it is easy to study. But it turns out that it is also very complex. It protects us from sun damage, the weather, and it has to constantly renew itself. Adult stem cells on the base of the epidermis divide to renew themselves and create new keratinocytes to migrate to the surface to replace those that are damaged or shed.”
If doctors deliver the missing gene, or even the protein it encodes, to those stem cells, it may help tie the epidermis and dermis together to prevent the blistering, skin loss and possibly even cancer. (Simply delivering it to mature keratinocytes is not enough because of the continual sloughing of the mature cells that occurs even in healthy skin.) But it’s been a journey of 20-plus years to move from concept to human trials.
“For us, it’s not enough to simply understand the molecular biology of the disease,” says Khavari. The team has had to move through a labyrinth of not just scientific challenges, but also the necessary regulatory approvals, from the institutional review boards that approve research involving human subjects to the Food and Drug Administration. They found themselves balancing their desire for speed with the need to cross every “t” and dot every “i” to ensure they would not inadvertently do more harm than good to the people they were trying so desperately to help.
Epidermolysis bullosa was first discovered in the late 1800s. It’s a member of a family of conditions called blistering diseases. EB occurs in three forms: simplex, junctional and dystrophic. Each of the three is caused by mutations affecting essential basement membrane components: the proteins keratin, laminin or collagen.
Junctional dystrophic EB is the most severe form of the disorder; these children lack laminin-332 and rarely live past 5 years of age. Recessive dystrophic EB, the form that Spaulding and Martinez have, is the next most severe. People with EB simplex can live relatively normal lives, although their skin blisters frequently, often on the hands and feet.

Stanford’s push to find a treatment or cure for EB can be traced back to 1988, when Bauer came to Stanford from Washington University in St. Louis. After completing an internship in dermatology Bauer became a postdoctoral scholar in Arthur Eisen’s laboratory at Washington University where he studied tadpole metamorphosis — how the animal loses its tail and grows legs to become an adult frog.
“One of the enzymes in this process was a protein called matrix metalloproteinase 1, or MMP1,” says Bauer. MMP1 degrades matrix proteins in the space between cells to allow the drastic tissue remodeling necessary to switch from wriggling to hopping. Although the role of type-7 collagen hadn’t yet been identified in EB, Bauer, who had continued to see the occasional dermatology patient during his postdoc years, began to wonder whether MMP1 could be responsible for the blistering seen in skin diseases like epidermolysis bullosa.
“You might say I moved from tadpoles to humans,” says Bauer. “I transferred back to the clinic in the early ’70s and began to focus my research increasingly on patients with epidermolysis bullosa.” He also developed a close relationship with Lynn and Gary Anderson, whose two children, Chuck and Christine, died from EB: Chuck, at 27, in 1988 and Christine, at 14, in 1993.
The Andersons launched the Los Angeles-based EB Medical Research Foundation in 1991 to raise funds to combat the disease. To date, the organization has raised over $5 million for EB research. “I cared for both Chuck and Christine and I consider Lynn and Gary to be personal friends,” says Bauer. “When you get to know patients and their families who are at first intimately affected, and then intimately committed and involved in the research effort, there’s a real potency and urgency brought to the table.”
That urgency translated into a cascade of research findings that led to the current clinical trial.
In the late 1980s, Robert Burgeson, PhD, and his research group at Shriner’s Hospital in Portland, Oregon, discovered type-7 collagen and helped show that patients with recessive dystrophic EB lacked this protein. In 1988, Bauer kept the ball rolling when he moved from St. Louis to Stanford to serve as the chair of dermatology.
“From 1988 to the early ’90s, we focused on identifying the mutations in the type-7 collagen gene that caused the disease, in the hopes that we could supply protein corrective therapy as had been done in other diseases,” says Bauer. “In the early to mid-1990s we were investigating whether it was possible to insert genes into cells, and turn them off and on. All these things were, of course, very rudimentary compared with what we are doing now.”
Khavari joined the team in 1993 after earning a PhD in the Stanford laboratory of Gerald Crabtree, PhD.
“Paul’s fundamental knowledge and skills in molecular biology techniques took our efforts to an entirely different level,” says Bauer.
Marinkovich, in turn, joined the team in 1995 after completing his fellowship training in the Burgeson laboratory. There, he had helped discover the laminin-332 protein and showed that it is defective or non-functional in children with the junctional dystrophic form of the disease. He’d also learned how this laminin works together with type-7 collagen to keep skin from blistering.
“Understanding how these proteins work together, and how this interaction is disrupted in EB, was key to our subsequent therapeutic studies,” says Marinkovich.
In 1995, Bauer became dean of the medical school and transferred his grants and the chairmanship of the department to Lane. But work continued apace.
“In 1996, a year before Garrett was born, we used gene therapy to correct a different genetic mutation in human skin cells and grow the then-normal skin on the backs of laboratory mice,” says Khavari. “This was the first time that the idea of correcting mutations in skin cells was shown to be possible.”
During this time, the doctors were often reminded of the awful toll of the disease. Khavari recalls a Halloween party for patients and staff hosted at Lane’s house about 10 years ago. “One child came in costume as a mummy, covered in bandages. During the evening, some of the blood from his wounds began to seep through the gauze, and those who didn’t know this child had EB were remarking about how realistic the bloodstains were. It’s impossible for me to imagine what it’s like to be the parent of that child.”
As Garrett grew, Montello developed what she called a “patch-and-go” philosophy, determined to give her son as normal a life as possible. When he turned 8 he got a bike for Christmas and rode it with abandon, disregarding his many bandages. “He’s such a fighter, and has developed such a positive attitude,” says Montello. “When he learned that kids with EB were sometimes called ‘butterfly kids’ he didn’t like it. He thought it was kind of girly. He said, ‘Can you call me a dragonfly kid instead?’ And that describes him perfectly. He is a bit of a dragon.”
Many EB children and their families adopt similar approaches.
“There’s something about these patients and family members that is very unique,” says Tang. “They seem to have a unique ability to accept the condition, and move on to what needs to happen to live the best life possible. It’s sad, but also so inspirational. What is it about the human spirit that keeps them going?”
Many of today’s gene therapy approaches rely on a class of viruses called retroviruses. In the normal course of their reproductive cycle, these viruses infect cells and insert their genetic material directly into those cells’ DNA until they’re ready to spread further. When using retroviruses for gene therapy, researchers remove disease-causing genes and sneak in an additional one — a gene that makes a missing or malfunctioning protein. Once the viruses insert the gene into the DNA of the patient’s cells, the cells can begin to make the missing protein.
Preparations for the current clinical trial began in earnest in 2003. In the first four years, the researchers conducted laboratory and animal studies to show the approach was technically feasible. From 2007 to 2010, they designed a virus capable of inserting the corrected type-7 collagen gene into a target cell’s genome so it could direct the creation of a functional version of the protein. This was hard in part because the gene is large and difficult to fit into the viral envelope.
Once the virus was made, the researchers had to find a way to manufacture it for use in humans. This required what the FDA has designated as “Current Good Manufacturing Practices,” or CGMP. Because Stanford did not have a CGMP facility, the researchers partnered with colleagues at Indiana University to use theirs. (Stanford is in the process of opening its own CGMP facility later this year.) They also had to overcome an alphabet soup of regulatory hurdles before a single patient could be treated.
“Fundamentally, the Stanford group has done something that no other EB research group in the world has yet done,” says Bauer, who is now the Lucy Becker Professor in Medicine, Emeritus. “It’s a wholly professional and rigorous approach by a spectacular group of people all focused on a single endpoint: to help patients with terrible lives. I’m very proud of them.”
In 2010, the team began to enroll five patients for the first trial. All of these first patients are adults, willing and able to give informed consent for their participation. (Trials involving children are subject to more stringent requirements.) Any future phases of the trial will likely include minors.
“Once we learn whether the treatment is safe, we’d like to include children as quickly as possible, in an attempt to improve their quality of life and prevent ongoing damage caused by scarring,” says Khavari.
The researchers need roughly 28 days to prepare for the experimental skin grafting. It’s conducted primarily in a specialized clean room on the second floor of the Lorry I. Lokey Stem Cell Research Building. The dimly lit room is small, with carefully controlled airflow and filters to remove any contaminants. Steps sound sticky as scientists walk over an adhesive mat to clean their shoes.
“The incubator is there,” says Siprashvili, indicating a portion of the room blocked off from the door by a transparent plastic curtain hanging down in flaps like the panels covering a car wash entrance. The tan metal box is about the size of a refrigerator, with two doors. It’s an unprepossessing repository of the hopes, dreams and years of hard work of patients and clinicians. Currently it shelters the genetically corrected cells of the fifth (and last) participant in the phase-1 clinical trial.
Once, the cells of the patient didn’t make type-7 collagen. Now they do.
Siprashvili supervises the care and feeding of the tiny plugs of skin cells obtained from the participants. Each of the subjects has recessive dystrophic EB. Four have now received grafts made of sheets of their own corrected skin cells, and researchers are preparing the skin grafts for the fifth. And then the researchers will watch, and wait.
Phase-1 clinical trials are meant to assess the safety of a particular intervention, not its efficacy. The patients will return for monitoring, at first on a monthly basis. Then if no adverse effects are seen (a loss of the skin graft, a propensity of the graft to migrate or to become cancerous, for example), the researchers will move on to a phase-2 trial, which they hope will include children. A phase-2 trial includes a larger number of patients and tests the effectiveness of a treatment along with its safety.
“This itself has been a 10-year process to get to this stage,” says Siprashvili. “We had to first create and outfit this room. And then we had to learn how to reliably and safely not only grow the cells from the patients, but also to design a virus to safely carry the corrected collagen gene into the cells.”
In layman’s terms, the process goes something like this: Collect two, 8-millimeter-square skin biopsies from patients; isolate the keratinocytes, which will contain epidermal stem cells as well; infect the cells with the genetically engineered virus carrying the corrected type-7 collagen gene; grow the cells over a period of about three weeks into eight thin sheets (each roughly the size of a playing card); conduct a series of tests on the cells to ensure their purity and safety; and then carefully, oh so carefully (!), hand-carry the newly minted skin in a small, air-tight box to the operating room where the patient waits under general anesthesia.
“Once the sheets of cells are grown, they have about a 24-hour half-life during which they must be grafted,” says Siprashvili. “As far as a production process for a biological product for use in humans, this is one of the most complex and difficult in the world.”
Each patient gets six grafts — five over previously existing wounds and one on a wound created deliberately during the grafting process. The patient spends a few days in the hospital, moving as little as possible to avoid disturbing the grafts before they’ve adhered to the underlying tissue.
A key part of the study is confirming that type-7 collagen is correctly incorporated into the anchoring fibrils that hold the skin layers together, and that the protein is fully functional.
“In terms of therapy, understanding the type-7 collagen protein has been just as critical as understanding the type-7 collagen gene,” says Marinkovich.
The first patient received the grafts in November 2013. Martinez, a long-standing patient of Marinkovich’s, was the third patient to participate in the trial. He received his grafts on Dec. 1, 2014: one graft on the top of his left hand, two on the top and side of his right hand, two on the side of his left foot, and one near his right heel.
“I don’t know how, or whether, my participation in this trial will benefit me, personally,” says Martinez, who lives alone but requires daily care. Unlike many EB patients, Martinez finished high school and went on to complete a degree in business marketing at a community college. But as the years pass, the disease is taking its toll. His hands are fused, and he no longer holds a job.
“I’m 32 years old, which is quite old for this disease,” says Martinez. “I’m lucky to be alive. I didn’t do this for myself; I did it for the children. I don’t want anyone to go through this. We have to stop this.”
It’s too early to tell whether the skin grafts will help any of the first participants. That’s not even the point of the trial. But physicians and patients are hopeful that grafting even small patches of tissue will make a difference in the recipients’ quality of life — perhaps by preventing blistering on especially vulnerable areas of the body, or slowing or stopping the development of mitten hand, or even helping chronic wounds heal at last.
“We need to not let ‘perfect’ be the enemy of ‘good enough,’” says Tang. “Even just preserving their hand and finger function would be a tremendous step forward.”
Still, Martinez feels that for him, for now, the results have been positive. His grafted skin has not blistered and appears relatively normal.
In May, the team presented preliminary evidence at the annual conference of the Society for Investigative Dermatology that, for the first three patients, the grafted skin expressed type-7 collagen at the basement membrane junction and appeared to have a normal number of anchoring fibrils across the dermis and epidermis.
“Even though my grafts are only the size of a playing card, the results have been life-changing,” says Martinez. “If you can minimize skin breakage, you minimize pain in that area, you minimize the chance of skin cancer in that area. It may not seem like a big deal, but this has really improved my quality of life.”
Spaulding was not a participant in the phase-1 trial. For one thing, he wasn’t yet an adult when the trial was launched. For another, he may not have the “right” mutation in his type-7 collagen gene.
Researchers have found dozens of mutations that can lead to EB. To enter the trial, patients had to have a particular type of mutation near the beginning of the gene. These patients would make just a small portion of the type-7 collagen protein. Exposure to this bit of the protein may reduce the chance that the patient’s immune system will react to, and try to destroy, the full-length protein made by the corrected gene.
“Garrett’s biopsy was inconclusive,” says Montello. “It’s possible that he may have an extra mutation that may preclude him from the trial. But we’re going to check again.”
Meanwhile, life goes on.
“I’ve been working with these patients since the early ’90s,” says Marinkovich. “Yet, I’m still affected every time I see these patients and their families. I could in no way do what they do. They overcome so much just by getting up in the morning. The types of obstacles I face in my life and my research seem minimal in comparison.”
Pain management is always a challenge, of course, with patients attempting to balance the need for comfort with the side effects of medications.
“I have a very high tolerance, and don’t take any pain medication,” says Martinez. “I cherish my mind a lot. Rather than feel like a zombie, I prefer to feel the pain and feel alive.”
Stanford’s EB team hopes to publish preliminary results of the phase-1 clinical trial this summer, but they’ll continue to monitor the impact on the participants for the rest of their lives. Like their patients and their patients’ parents, they are elated that the research has finally progressed this far.
“In the seven years since Jackson was born, we’ve seen tremendous progress,” says Jamie Silver, of the fundraising and advocacy organization, EB Research Partnership, whose son has EB. “This idea of gene therapy for EB wasn’t a reality at the time. It was in the lab, but it wasn’t anywhere near a human. Now, at Stanford, it is in clinical trials. We are seeing tangible progress. That gives us as parents and us as an organization the drive to keep going.”
Spaulding is now a studious young man with a shy, sweet smile and a passion for Apple products. Confined now to a wheelchair, he looks much younger than his 18 years. He struggles with kidney problems and last year he had all his teeth removed because of ongoing dental issues. He’s recently assumed the CEO position in a newly formed family business selling artisan olive oil and vinegars — a position he juggles along with his Advanced Placement classes as a junior at Orestimba High School, in Newman, California. His family started the business when Spaulding expressed a desire to make an impact with his life, however short it may be.
Not surprisingly, some of his doctors were also his first customers.
“They are some of my best friends,” says Spaulding, who has, of course, known many of them all his life. “Dr. Lane talks with me on the phone, and they are always keeping me up-to-date on the latest advances. Their research has already definitely helped me. Some of the new bandaging and skin care techniques have really made a huge difference.” Aggressive bandaging has kept his fingers separated and maintained his hand function — the better to use the new iPad he received from the Make-A-Wish Foundation to do homework and run his business, Montello Fine Foods.
When asked about the ongoing clinical trial, Spaulding is noncommittal, expressing a concern that participation in a future phase could interfere with his school work. But, with his trademark forward-thinking attitude, what he really wants to talk about is olive oil, balsamic vinegar and recipes.
“When he had his teeth removed, I was worried about him,” says Montello. “Here we were starting a business based entirely on food, and he can’t eat. About two weeks after the surgery, I asked him, ‘How are you doing, Garrett? How are you really doing? Are you still the happy little camper I remember, or is this getting to be too much?’ And he looked at me, and smiled that smile, and said, ‘You know, Mom, as long as things don’t get any worse, I wouldn’t mind living to be 100.’”
Related Stanford Medicine magazine content:
- January 2023: Healing gel: Dramatic experimental gene therapy advance for blistering disease wound care. Related video: New gene therapy gel heals painful skin disease | 90 Seconds w/ Lisa Kim
Related Stanford News Center content:
- December 2022: Gel treatment heals blistering wounds. Related video: New gene therapy gel heals painful skin disease | 90 Seconds w/ Lisa Kim
- March 2022: Gene-therapy gel shows promise for skin disease
- November 2016: Gene therapy for blistering skin disease
- November 2014: Correcting a devastating collagen defect
