
How cells self-destruct
Discoveries about the ways cells die are inspiring new disease treatments
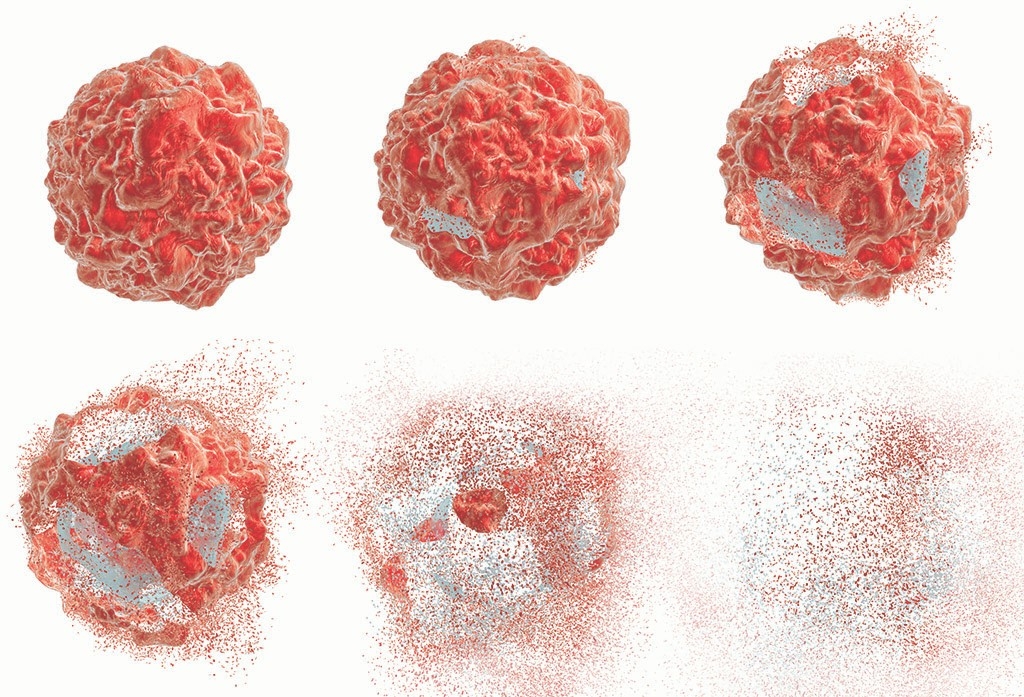
Inside every sick person, there’s a microscopic scene of destruction. Viruses, simple but nefarious lurkers, commandeer healthy cells to breed infection that, unchecked, would overrun the body.
But the body is equipped with weapons of its own. Like a bursting supernova, the infected cell can explode from the inside out. Its guts spew, and “alert molecules” are flung from the eruption, sending a message to neighboring cells: Danger is nigh.
The process is known as necroptosis, one of the more recently discovered forms of cell death, and it’s crucial to foiling viral enemies.
The best understood form of cell death — a self-destruct routine called apoptosis — was identified in 1972. Since then, scientists have sleuthed out several more highly controlled cell suicide processes that protect the body from pathogenic threats. But, as with any complex process, there are flaws. Too little cell death can contribute to diseases like cancer, while too mucx`h can promote autoimmune disease.
Related reading
A look at all the ways your cells die.
As scientists learn more about the chemical interactions inside cells that lead to cell death, some are using that knowledge for therapeutics by inducing cell suicide to kill tumor cells or repressing it to temper rheumatoid arthritis and multiple sclerosis.
“It’s exciting to think about how we can induce cell death in the proper way to fend off or stunt illness,” said Jan Carette, PhD, an associate professor of microbiology and immunology at Stanford who investigates cell death and whose most recent research has zeroed in on necroptosis. “But first we need to understand how these modes of death occur, how they differ and how they intersect. That’s what will drive therapeutics.”
In the beginning, there was one
Before apoptosis was discovered, many scientists believed that cells died only by accident or injury, expiring in a disorganized catastrophe. Observations of the tightly orchestrated molecular interactions that take place during apoptosis proved that wrong. The key to the process is a family of molecules called caspases, which act as shepherds of cell death and activate processes such as DNA degradation and the breakdown of organelles in the cell.
Like most other forms of cell death, apoptosis is a mediator of health. It’s switched on to rid the body of cells that are cancerous or infected, or of cells that don’t form properly, among other duties. But apoptosis is not exclusively for expunging dysfunctional cells; it’s also active during embryo development, sculpting fingers and toes from the paddlelike ends of our early extremities.
Once apoptosis was recognized, researchers lumped all other forms of cell death into a category they called “necrosis,” which was loosely comparable to a shoulder shrug: They knew cells could die in ways other than apoptosis, but what those were was still a mystery. Today, that catchall category has been spliced and spliced again, making room for a spectrum of microscopic deaths.
Necroptosis, the explosive response to infection, is one of these relatively newly recognized types of cell death. Though it primarily plays out during infection, too little necroptosis has also been implicated in cancer, whereas too much instigates inflammation-based autoimmune diseases, such as irritable bowel syndrome and rheumatoid arthritis.
Necroptosis was discovered and named in 2005 when Junying Yuan, PhD, a seasoned cell-death scientist and professor of cell biology at Harvard University, experimentally disabled the apoptotic pathway in a cell. The cell was then put under lethal conditions and, much to the scientist’s surprise, it died by a process that looked nothing like apoptosis. Robbed of its original exit strategy, the cell had initiated a backup plan.
“The concept of another kind of programmed cell death was paradigm-shifting because, so far, apoptosis had owned the exclusive rights.”
“The concept of another kind of programmed cell death was paradigm-shifting because, so far, apoptosis had owned the exclusive rights,” said Carette. Yuan’s discovery meant apoptosis was not the only type of cell death implicated in both the protection against and the perpetuation of disease.
In 2011, when Carette established his lab at Stanford, cell death was nowhere on his radar. Instead he was developing a new technology to pinpoint genes important for specific biological functions. “At that time, the necroptosis field was just starting — almost nothing was known about the pathway that leads to this potent form of death,” said Carette. While looking for biological functions to study, he and his lab members attended a talk about the necroptosis process and saw an opening.
“When they described the pathway, there were all these question marks on their slides where the names of genes should have been,” said Carette. He and his lab members had a hunch that their technology could fill in those question marks. And so Carette’s new quest began.
Back to the basics
No matter whether necroptosis is acting as a defender against disease or as a perpetrator of illness, its underlying mechanism is the same. The killing blow is delivered when something Carette refers to as “the executioner protein” stirs from dormancy. This protein, formally known as MLKL, has a sort of Bruce Banner-turned-Hulk quality to it — docile when off duty, but extremely hostile when riled up.
“We found it odd that human cells carry this potent molecule that, once activated, literally kills cells from within,” said Carette. “So the question became, what keeps this weapon silent when there is no danger, and what prods it into ‘killer’ mode?”
The unleashing of MLKL is preceded by a whole cascade of signals and lock-and-key steps, many of which Carette and his team have helped discover. The protein then bores through the cell’s covering and bursts out, exploding the cell like a water balloon speared by a needle from within. The cell’s insides, including special signaling molecules, gush into the open. The signaling molecules’ message to the cells they encounter: Gird yourself or prepare for death. Depending on the severity of infection or disease, those cells will follow suit and explode, perpetuating the cycle.
“We found it odd that human cells carry this potent molecule that, once activated, literally kills cells from within. So the question became, what keeps this weapon silent when there is no danger, and what prods it into ‘killer’ mode?”
Just this past year, Carette and his group discovered a unique and critical detail about the activation of MLKL: It requires a code. This “death code,” as Carette coined it, consists of a string of specific molecules that latches onto MLKL in the final phase of activation to set it loose. Without it, MLKL is harmless.
“This is something that no one knew about, or really had even thought about before,” said Cole Dovey, PhD, a postdoctoral scholar in Carette’s lab. “We’re really excited about the possibility of leveraging this information to develop targeted, precise therapies for certain diseases in which necroptosis is a main culprit.” Dovey is hopeful that future work employing the “death code” might help tame inflammatory or autoimmune diseases by blocking the code from MLKL to prevent unprovoked necroptosis.
Already, pharmaceutical companies are beginning to capitalize on this idea, developing drugs that inhibit necroptosis to treat irritable bowel syndrome and rheumatoid arthritis. On the flip side, inducing necroptosis in diseased cells is an attractive tactic. While scientists have succeeded in triggering apoptosis in some cancers, many are now toying with necroptosis activation in the hopes that it can add to a growing arsenal of immune-based therapies.
The next cell death
As necroptosis continues to evolve in its therapeutic utility, other programmed death pathways are coming into their own. The latest, ferroptosis, was discovered in 2012 by Scott Dixon, PhD, then a postdoctoral scholar, and his colleagues in the laboratory of Brent Stockwell, PhD, professor of biological science and chemistry, at Columbia University. How it works is still obscure.
“Ferroptosis just hasn’t been on our radar that long,” said Dixon, now an assistant professor of biology at Stanford. “Scientists have known about necroptosis for 13 years and apoptosis for more than 40, so there’s been a lot more time for research to get underway.”
After coming across other non-apoptotic cell deaths like necroptosis, scientists suspected there were more, and continued to search. That’s how Dixon and Stockwell discovered ferroptosis, an iron-dependent demise unique from any found before.
Now Dixon investigates the drivers behind ferroptosis and the hand it plays in human disease. For now, scientists know that iron molecules help propel ferroptosis, and that it occurs when a particular protein building block, cysteine, and a compound called glutathione are dually low in the cell. Both cysteine and glutathione are critical to a process that wipes out destructive molecules called reactive oxygen species.
“We knew how to induce ferroptosis in a dish, and we thought if we could find inhibitors of the process, we could track down where ferroptosis was biologically important.”
Overall, though, ferroptosis’ role in health and disease is still opaque, which is in part because of some technical limitations. Both apoptosis and necroptosis exhibit molecular markers — evidence that a biological process has occurred or is present, like “signature” proteins or molecules that are unique to specific pathways. That’s not the case when ferroptosis has been at work.
“Let’s say you wanted to look at a postmortem brain and see if ferroptosis contributed to that death. Right now, there’s no way to inspect a section of the tissue to see if those cells indeed died through ferroptosis,” said Dixon. “There’s just no marker. Not yet at least.”
So when they discovered ferroptosis, Dixon and his lab colleagues had to find a workaround to be able to spot ferroptosis in action.
“We knew how to induce ferroptosis in a dish, and we thought if we could find inhibitors of the process, we could track down where ferroptosis was biologically important,” Dixon explained. So they screened an enormous set of molecules for their ability to stop the new death pathway, and selected a handful that seemed to halt progress.
Dixon’s lab at Stanford is still focused on the fundamentals, continuing to decipher ferroptosis’ pathway, but scientists outside of his lab have been able to use the inhibitors he discovered to understand how ferroptosis fits into the bigger picture of health. “You can take these inhibitors and test them in different models of disease — stroke, Parkinson’s, Alzheimer’s and Huntington’s, among others — in various organisms,” said Dixon. “And in many cases, it’s turning out that the inhibitors we have found protect the models from cell death and subsequent damage.”
For Dixon and Carette, one of the next big questions is understanding exactly how the various microscopic deaths intertwine. There’s already evidence to suggest that apoptosis and necroptosis somehow act as backup mechanisms for each other — if a pathogen thwarts necroptosis, apoptosis could theoretically pick up the slack.
“There are a few tentative reports that draw connections between ferroptosis, necroptosis and apoptosis, but the data are scattered,” said Dixon. “Everyone is trying to forge those links, but I don’t think anybody really knows yet. That’s going to be the next frontier.”
