
COVID’s unwitting enablers?
New research flags unexpected cells in lungs as a suspected source of severe COVID
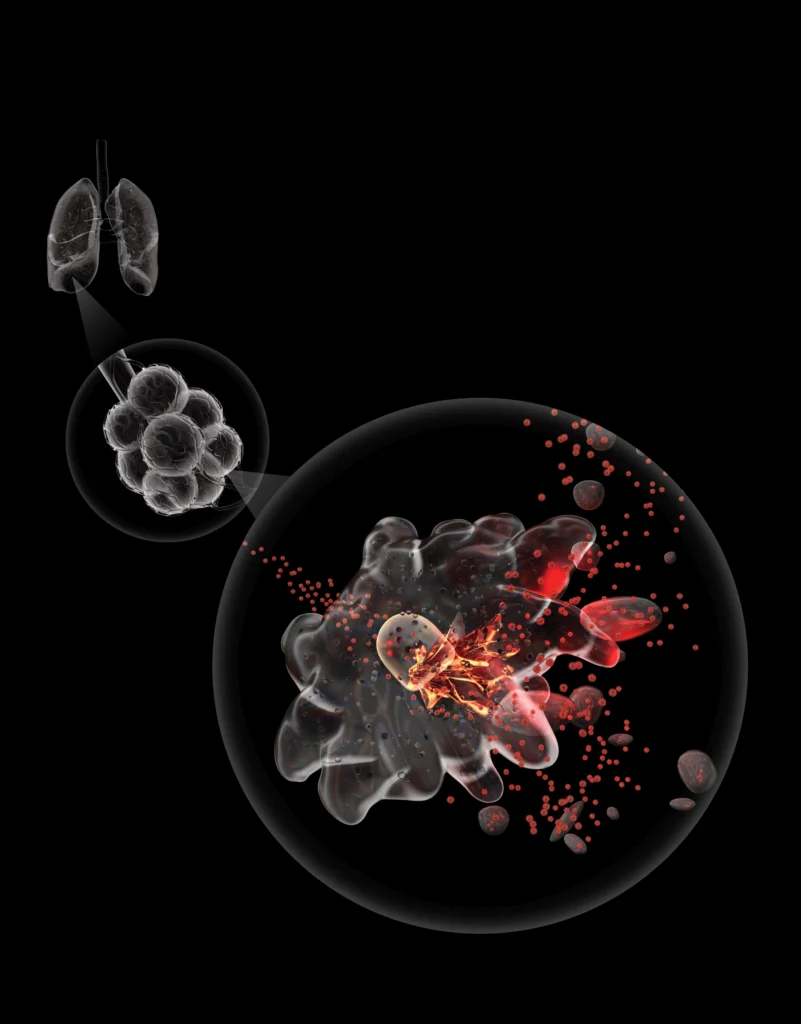
Mild COVID is manageable. Severe COVID can kill you. If we knew why a mild case turns into a severe one, it might help.
We could start with a look at your lungs, whose surface is squiggly, uneven terrain. That’s because it’s covered with minuscule, air-filled bubbles called alveoli. There are about 500 million alveoli in a pair of healthy adult lungs. The wall enclosing each tiny air pocket is just one cell thick.
Your lungs’ bubbly contour makes their total surface area huge. If it could be spread out to form a flat sheet, that surface would coat the entire floor of a tennis court. That’s great for the transfer of oxygen from the air you’ve just inhaled to the red blood cells that will carry their vital cargo to destinations throughout your body.
But that permeable surface’s 1/100,000-inch thickness also makes it the most fragile of tissues: a mostly one-cell-thick border through which microbial pathogens can burrow, ride to their favorite targets and maybe, on the way, feast on the nutrient-rich buffet on offer in your bloodstream. Should anything — infection, inflammation or injury — disrupt that layer of lung-surface cells, what’s on one side could leak out and what’s on the other side could get in. Not good for your health.
That diaphanous border, however, doesn’t go unpoliced. Specialized immune cells patrol both sides of it, scarfing down unwanted intruders and calling up reinforcements on active patrol nearby or snoozing in our bodies’ barracks. We wouldn’t last long without them.
Here’s the thing: We’re finding out that the road to severe COVID may be paved with lung-resident immune cells we thought were our friends. And all the while, we’ve been pointing an accusing finger at other cells in our lungs that — although, OK, they may not exactly be perfectly innocent — are, relatively speaking, rank amateurs.
We’re finding out that the road to severe COVID may be paved with lung-resident immune cells we thought were our friends.
A type of immune cell known as an interstitial macrophage has recently been implicated in the critical transition from a merely bothersome COVID case to a potentially deadly one. Interstitial macrophages are situated deep in the lungs, ordinarily protecting that precious organ by, among other things, devouring viruses, bacteria, fungi and dust particles that make their way down our airways.
But as Stanford Medicine researchers showed in a study published in April in the Journal of Experimental Medicine, it’s these very cells that, of all known types of cells composing lung tissue, are most susceptible to infection by SARS-CoV-2.
SARS-CoV-2-infected interstitial macrophages, the scientists have learned, once infected, can squirt out inflammatory and scar-tissue-inducing chemical signals, potentially paving the road to pneumonia and damaging the lungs to the point where the virus, along with those potent secreted substances, can break out of the lungs and wreak havoc throughout the body.
The surprising findings point to brand-new approaches to preventing a SARS-CoV-2 infection from stepping over the line beyond which a manageable disease becomes a life-threatening one. Indeed, they may explain why precisely targeted drugs called monoclonal antibodies meant to combat severe COVID didn’t work well, if at all. When they did work, it was only when they were administered early in the course of infection, when the virus was infecting cells in the upper airways leading to the lungs but hadn’t yet ensconced itself deep in lung tissue.
Monoclonal antibodies are designed to bind strongly to this or that specific feature on the surface of an invading virus, with the usual objective of blocking its ability to bind to its receptor on a target cell’s surface. But when a “this” becomes a “that” because of a mutation that changes the shape of the viral surface, the antibody is out of a job, and a new one must be designed.
The virus surprises
“We’ve overturned a number of false assumptions about how the virus actually replicates in the human lung,” said Catherine Blish, MD, PhD, the George E. and Lucy Becker Professor in Medicine II and associate dean for basic and translational research.
Blish is the co-senior author of the study, along with Mark Krasnow, MD, PhD, the Paul and Mildred Berg Professor and Executive Director of the Vera Moulton Wall Center for pulmonary vascular disease.
“The critical step, we think, is when the virus infects interstitial macrophages, triggering a massive inflammatory reaction that can flood the lungs and spread infection and inflammation to other organs,” said Krasnow, a professor of biochemistry. Blocking that step, he said, could prove to be a major therapeutic advance. But there’s a plot twist: The virus has an unusual way of getting inside these cells — a route drug developers have not yet learned how to block effectively — necessitating a new focus on that alternative mechanism, he added.
In a paper published in Nature in early 2020, Krasnow and his colleagues described a technique they’d worked out for isolating cells from fresh human lung tissue; dissociating the cells from one another; and characterizing them, one by one, on the basis of which genes within each cell were active and how much so. Using that technique, the Krasnow lab and collaborators were able to discern more than 50 distinct cell types, assembling an atlas of healthy lung cells.
“We can’t say that a lung cell sitting in a dish is going to get COVID. But we suspect this may be the point where, in an actual patient, the infection transitions from manageable to severe.”
Catherine Blish, MD, PhD, associate dean for basic and translational research.
“We’d just compiled this atlas when the COVID-19 pandemic hit,” Krasnow said. Soon afterward, he learned that Blish and Arjun Rustagi, MD, PhD, then an instructor of infectious diseases, were building an ultra-safe facility where they could safely grow SARS-CoV-2 and infect cells with it.
A collaboration ensued. Krasnow and Blish and their associates obtained fresh healthy lung tissue excised from seven surgical patients and five deceased organ donors whose lungs were virus-free but weren’t used in transplants. After infecting the lung tissue with SARS-CoV-2 and waiting one to three days for the infection to spread, the researchers separated and typed the cells to generate an infected-lung-cell atlas, analogous to the one Krasnow’s team had created with healthy lung cells. They saw most of the cell types that Krasnow’s team had identified in healthy lung tissue.
Now the scientists could compare pristine versus SARS-CoV-2-infected lung cells of the same cell type with one another: They wanted to know which cells the virus infected, how easily SARS-CoV-2 replicated in infected cells, and which genes the infected cells cranked up or dialed down compared with their healthy counterparts’ activity levels. They did this for each of the dozens of cell types they’d identified in both healthy and infected lungs.
“It was a straightforward experiment, and the questions we were asking were obvious,” Krasnow said. “It was the answers we weren’t prepared for.”
Where air meets blood
The cells the researchers had expected to succumb most readily and ominously weren’t the ones that did.
It’s been assumed that the cells in the lungs that are most vulnerable to SARS-CoV-2 infection are those known as alveolar type 2 cells. That’s because the surfaces of these cells, along with those of numerous other cell types in the heart, gut and other organs, sport many copies of a molecule known as ACE2. SARS-CoV-2 has been shown to be able to grab onto ACE2 and manipulate it in a way that allows the virus to maneuver its way into cells.
Alveolar type 2 cells are somewhat vulnerable to SARS-CoV-2, the scientists confirmed. But the cell types that were by far the most frequently infected turned out to be two varieties of a cell type called a macrophage.
The word “macrophage” comes from two Greek terms meaning, roughly, “big eater.” This name is not unearned. The air we inhale carries not only oxygen but also, unfortunately, tiny airborne dirt particles, fungal spores, bacteria and viruses to our lungs. A macrophage earns its keep by, among other things, gobbling up these foreign bodies.
The airways leading to our lungs culminate in myriad alveoli, which are abutted by abundant capillaries. This interface, called the interstitium, is where oxygen in the air we breathe enters the bloodstream and is then distributed to the rest of the body.
The two kinds of SARS-CoV-2-susceptible lung-associated macrophages are positioned in two different places. Cells known as alveolar macrophages hang out by the billions patrolling the inner surfaces of the alveoli. As expected, SARS-CoV-2 can infect alveolar macrophages. Once infected, these cells smolder, producing and dribbling out some viral progeny at a casual pace but more or less keeping a stiff upper lip and maintaining their normal function. This behavior may allow them to feed SARS-CoV-2’s progression by incubating and generating a steady supply of new viral particles that escape by stealth and penetrate the layer of cells enclosing the alveoli.
Interstitial macrophages, the cell type now revealed to also be infected by SARS-CoV-2, patrol the outer surface of the alveoli, where the rubber of oxygen meets the road of red blood cells. If an invading viral particle or other microbe manages to evade alveolar macrophages’ vigilance, infect and punch through the layer of cells enclosing the alveoli — jeopardizing not only the lungs but also the rest of the body — interstitial macrophages are ready to jump in and protect the neighborhood.
At least, usually. But when an interstitial macrophage meets SARS-CoV-2, it’s a different story. Rather than get eaten by the omnivorous immune cell, the virus infects it.
The devil’s spatula
An infected interstitial macrophage doesn’t just smolder; it catches on fire. The virus literally seizes the controls and takes over, hijacking the cell’s protein- and nucleic-acid-making machinery. In the course of producing massive numbers of copies of itself, SARS-CoV-2 destroys the boundaries separating the cell nucleus from the rest of the cell, like the devil’s spatula shattering, splattering and scattering the yolk of a raw egg. (This deformation is denoted by the hideous term “nuclear blebbing.”) The cell’s outer membrane explodes, allowing viral progeny to exit the spent macrophage and move on to mess up other cells.
But that’s not all. In contrast to alveolar macrophages, infected interstitial macrophages pump out substances that signal other immune cells elsewhere in the body to head for the lungs. In a patient, Krasnow suggested, this would trigger an inflammatory influx of such cells. As the lungs fill with cells and fluid that accompany inflammation, oxygen exchange becomes impossible. The barrier maintaining alveolar integrity grows progressively damaged. Leakage of infected fluids from damaged alveoli propels viral progeny into the bloodstream, blasting the infection and inflammation to distant organs.
Yet other substances released by SARS-CoV-2-infected interstitial macrophages stimulate the production of fibrous material in connective tissue, resulting in scarring of the lungs. In a living patient, the replacement of oxygen-permeable cells with scar tissue would further render the lungs incapable of executing oxygen exchange.
“We can’t say that a lung cell sitting in a dish is going to get COVID,” said Blish, a professor of infectious diseases and ofmicrobiology and immunology. “But we suspect this may be the point where, in an actual patient, the infection transitions from manageable to severe.”
Another point of entry
Compounding this unexpected finding is the discovery that SARS-CoV-2 uses a different route to infect interstitial macrophages than the one it uses to infect the other types.
While SARS-CoV-2 gains access to alveolar type 2 cells and alveolar macrophages by clinging to ACE2 receptors on those cells’ surfaces, the virus breaks into interstitial macrophages using a different receptor these cells display. In the study, blocking SARS-CoV-2’s binding to ACE2 protected the former cells but failed to dent the latter cells’ susceptibility to SARS-CoV-2 infection.
“SARS-CoV-2 was not using ACE2 to get into interstitial macrophages,” Krasnow said. “It enters via another receptor called CD209.”
That would seem to explain why monoclonal antibodies developed specifically to block SARS-CoV-2/ACE2 interaction failed to mitigate or prevent severe COVID cases. To keep SARS-CoV-2 from binding to the alternate receptor on interstitial macrophages, those monoclonals would have to be reconfigured to aim at a brand-new bull’s-eye.
It’s time to find a whole new set of drugs that can hit that bull’s-eye and impede SARS-CoV-2/CD209 binding. As in, pronto, Krasnow said.
Krasnow said he has heard from a potential European collaborator who’s developing molecules that block CD209 and would like to test these compounds’ capacity to prevent SARS-CoV-2 from binding to interstitial macrophages.
With COVID-19 cases once again on the uptick, that sounds like a good idea.
